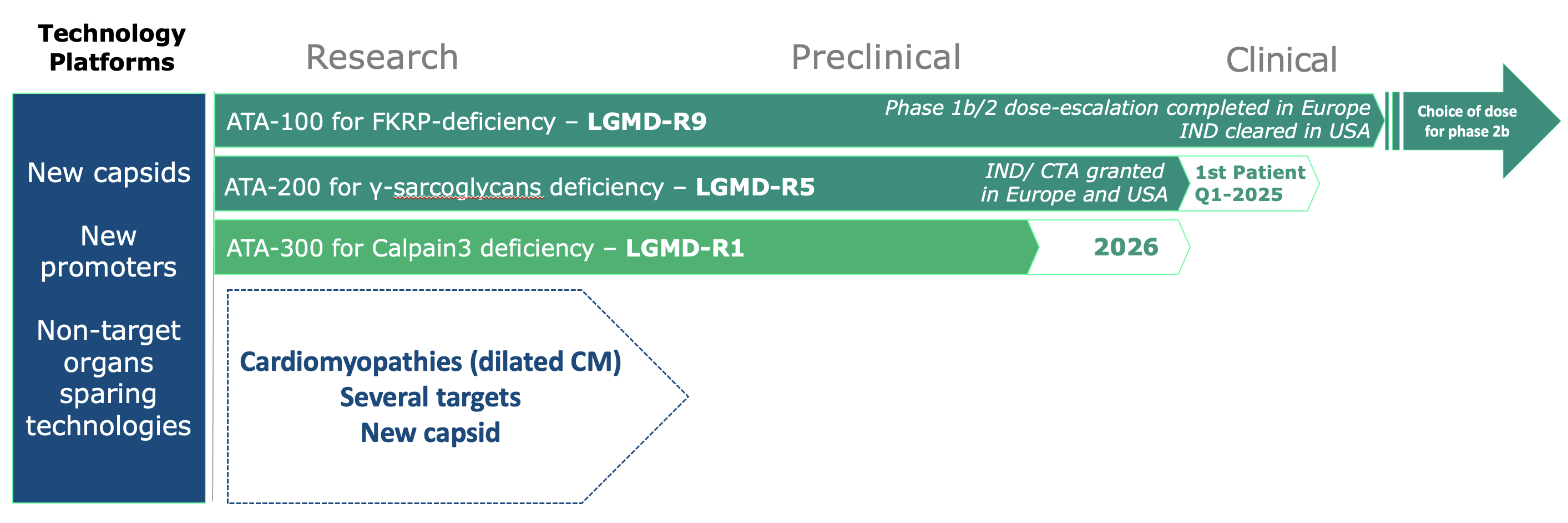
Our Pipeline
At Atamyo, we use our expertise in gene therapy to find new approaches for gene replacement.
We are focused on the development of a new generation of effective and safe gene therapies for muscular dystrophies and cardiomyopathies that currently lack treatment options.
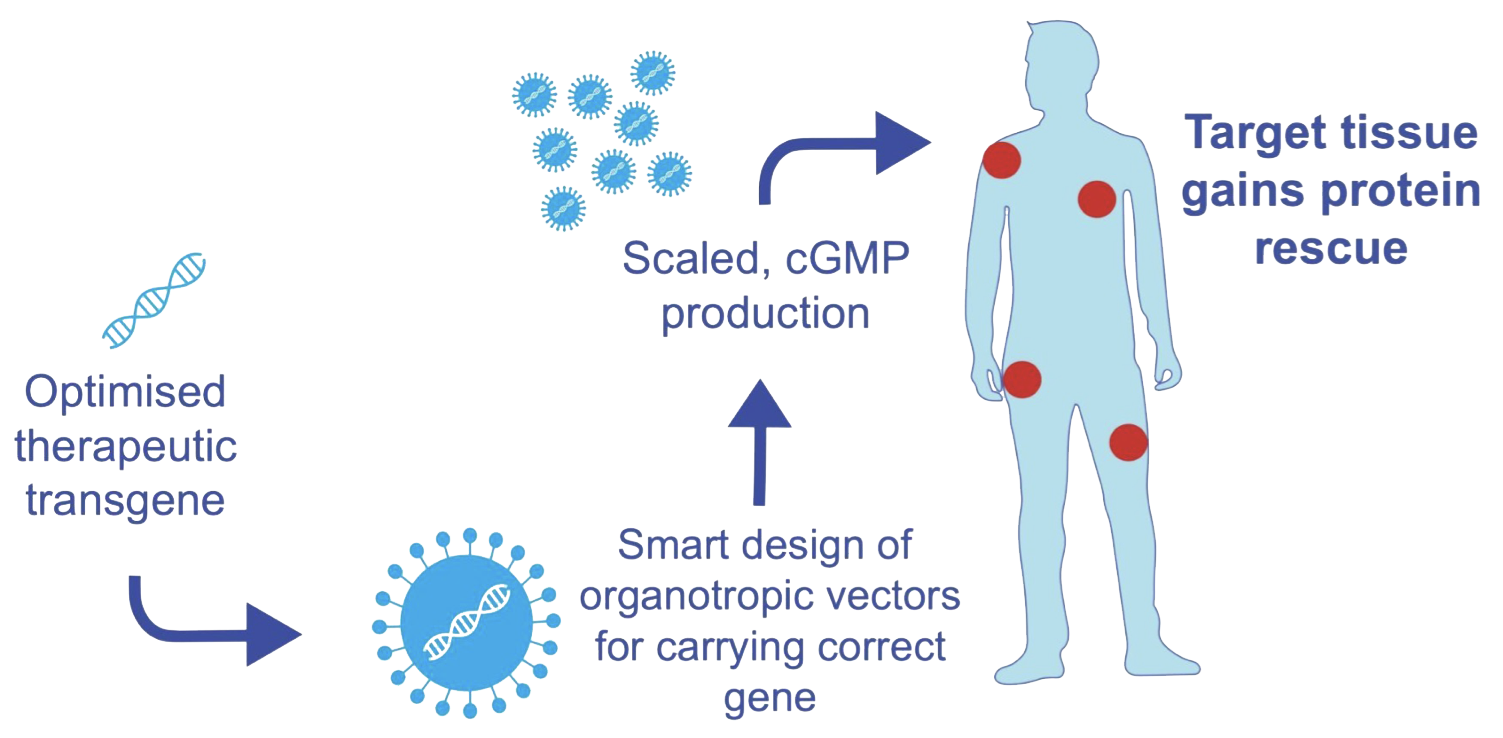
Development Programs
Our first clinical targets are monogenic Limb-Girdle Muscular Dystrophies (LGMDs), including LGMD R9, R5, and R1